


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Yamashita, Keishiro*; Nakayama, Kazuya*; Komatsu, Kazuki*; Ohara, Takashi; Munakata, Koji*; Hattori, Takanori; Sano, Asami; Kagi, Hiroyuki*
Acta Crystallographica Section B; Structural Science, Crystal Engineering and Materials (Internet), 79(5), p.414 - 426, 2023/10
Times Cited Count:0 Percentile:0.02(Chemistry, Multidisciplinary)The structure of a recently-found hyperhydrated form of sodium chloride, NaCl 13H(D)
13H(D) O, has been determined by
O, has been determined by  single-crystal neutron diffraction at 1.7 GPa and 298 K. It has large hydrogen-bond networks and some water molecules have distorted bonding features such as bifurcated hydrogen bonds and five-coordinated water molecules. The hydrogen-bond network has similarities to ice VI in terms of network topology and disordered hydrogen bonds. Assuming the equivalence of network components connected by pseudo symmetries, the overall network structure of this hydrate can be expressed by breaking it down into smaller structural units which correspond to the ice VI network structure. This hydrogen-bond network contains orientational disorder of water molecules in contrast to the known salt hydrates. Here, we present an example for further insights into a hydrogen-bond network containing ionic species.
single-crystal neutron diffraction at 1.7 GPa and 298 K. It has large hydrogen-bond networks and some water molecules have distorted bonding features such as bifurcated hydrogen bonds and five-coordinated water molecules. The hydrogen-bond network has similarities to ice VI in terms of network topology and disordered hydrogen bonds. Assuming the equivalence of network components connected by pseudo symmetries, the overall network structure of this hydrate can be expressed by breaking it down into smaller structural units which correspond to the ice VI network structure. This hydrogen-bond network contains orientational disorder of water molecules in contrast to the known salt hydrates. Here, we present an example for further insights into a hydrogen-bond network containing ionic species.
Kai, Takeshi; Toigawa, Tomohiro; Matsuya, Yusuke*; Hirata, Yuho; Tezuka, Tomoya*; Tsuchida, Hidetsugu*; Yokoya, Akinari*
RSC Advances (Internet), 13(11), p.7076 - 7086, 2023/03
Times Cited Count:3 Percentile:81.33(Chemistry, Multidisciplinary)Scientific insights of water radiolysis are widely used in the life sciences and so on, however, the formation mechanism of radicals, a product of water radiolysis, is still not well understood. We are challenging to develop a simulation code to solve this formation mechanism from the viewpoint of radiation physics. Our first-principles calculations have revealed that the behavior of secondary electrons in water is governed not only by collisional effects but also by polarization effects. Furthermore, from the predicted ratio of ionization to electronic excitation, based on the spatial distribution of secondary electrons, we successfully reproduce the initial yield of hydrated electrons predicted in terms of radiation chemistry. The code provides us a reasonable spatiotemporal connection from radiation physics to radiation chemistry. Our findings are expected to provide newly scientific insights for understanding the earliest stages of water radiolysis.
 - and K-montmorillonite
- and K-montmorilloniteKawakita, Ryohei; Saito, Akito*; Sakuma, Hiroshi*; Anraku, Sohtaro; Kikuchi, Ryosuke*; Otake, Tsubasa*; Sato, Tsutomu*
Applied Clay Science, 231, p.106722_1 - 106722_7, 2023/01
Times Cited Count:1 Percentile:21.06(Chemistry, Physical)Yotsuji, Kenji*; Tachi, Yukio; Sakuma, Hiroshi*; Kawamura, Katsuyuki*
Genshiryoku Bakkuendo Kenkyu (CD-ROM), 29(2), p.63 - 81, 2022/12
The understanding of the swelling phenomenon of montmorillonite is essential to predict the physical and chemical behavior of clay-based barriers in radioactive waste disposal systems. This study investigated the key factors controlling crystalline swelling behavior of montmorillonite with different interlayer counter-ions by molecular dynamics (MD) simulations. On the basis of the comparisons between MD simulated and experimental results, the water content in the interlayer in five homoionic (Na , K, Cs, Ca and Sr) montmorillonite was strongly correlated to the hydration number and the preference of an outer- or inner-sphere complex of each counter-ion. The detailed analysis for these results offer insights that the hydration number is controlled by the hydration free energy, the volume and the distribution of each interlayer counter-ion. The systematic MD simulations with virtually variable parameters clarified that the hydration free energy and the charge of interlayer counter- ions compete as influencing factors, and the control the formation rate of an outer-sphere complex of each counter-ion. The empirical relationships between these key factors will allow essential insights into predicting the swelling behavior of montmorillonite with different interlayer counter-ions.
, K, Cs, Ca and Sr) montmorillonite was strongly correlated to the hydration number and the preference of an outer- or inner-sphere complex of each counter-ion. The detailed analysis for these results offer insights that the hydration number is controlled by the hydration free energy, the volume and the distribution of each interlayer counter-ion. The systematic MD simulations with virtually variable parameters clarified that the hydration free energy and the charge of interlayer counter- ions compete as influencing factors, and the control the formation rate of an outer-sphere complex of each counter-ion. The empirical relationships between these key factors will allow essential insights into predicting the swelling behavior of montmorillonite with different interlayer counter-ions.
 FeO
FeO
Yagi, Yutaro*; Wakita, Yudai*; Kagomiya, Isao*; Matsue, Ikuya*; Kakimoto, Kenichi*; Matsumura, Daiju; Yoneda, Yasuhiro
ChemistrySelect (Internet), 7(21), p.e202104575_1 - e202104575_7, 2022/06
Times Cited Count:0 Percentile:0(Chemistry, Multidisciplinary)Kimoto, Kazushi*; Kawamura, Katsuyuki*; Makino, Hitoshi
Journal of Computer Chemistry, Japan, 19(2), p.46 - 49, 2020/00
This study proposes a 2D coarse-grained molecular dynamics (CGMD) method for the compaction simulation of montmorillonite clay. In the CGMD method, a unit structure of a water-hydrated clay molecule is coarse-grained into a particle. Thus, the deformable molecules are modeled as a set of linearly connected coarse-grained particles. As the inter-particle forces, the intra-molecular bonding and inter-molecular van der Waals forces are considered. For simplicity, the intra-molecular bonding is modeled as a linear harmonic oscillator, while the Lenard-Jones potential is used to define the van der Waals force field. With this model, the mechanical compaction of moistured montmorillonite is numerically simulated to find that 4-6 considerably deformed molecules are layered as a result of the compaction. It is alsofound that the simulated XRD pattern agrees to the experiment in terms of the peak angle.
 X-ray and neutron diffraction methods
X-ray and neutron diffraction methodsYamashita, Keishiro*; Komatsu, Kazuki*; Hattori, Takanori; Machida, Shinichi*; Kagi, Hiroyuki*
Acta Crystallographica Section C; Structural Chemistry (Internet), 75(12), p.1605 - 1612, 2019/12
Times Cited Count:7 Percentile:58.9(Chemistry, Multidisciplinary)A crystal structure of a high-pressure phase of magnesium chloride hexahydrate (MgCl 6HO-II) and its deuterated counterpart (MgCl 6DO-II) have been identified for the first time by in-situ single-crystal X-ray and powder neutron diffraction. The crystal structure was analyzed by the Rietveld method for the neutron diffraction pattern based on the initial structure determined by single-crystal X-ray diffraction. This high-pressure phase has a similar framework to that in the known ambient-pressure phase, but exhibits some structural changes with symmetry reduction caused by a subtle modification in the hydrogen-bond network around the Mg(HO)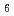 octahedra. These structural features reflect the strain in the high-pressure phases of MgCl hydrates.
octahedra. These structural features reflect the strain in the high-pressure phases of MgCl hydrates.
Fedkin, M. V.*; Shin, Y. K.*; Dasgupta, N.*; Yeon, J.*; Zhang, W.*; van Duin, D.*; Van Duin, A. C. T.*; Mori, Kento*; Fujiwara, Atsushi*; Machida, Masahiko; et al.
Journal of Physical Chemistry A, 123(10), p.2125 - 2141, 2019/03
Times Cited Count:44 Percentile:94.92(Chemistry, Physical)no abstracts in English
Matsuo, Tatsuhito; Arata, Toshiaki*; Oda, Toshiro*; Nakajima, Kenji; Kawamura, Seiko; Kikuchi, Tatsuya; Fujiwara, Satoru
Biochemistry and Biophysics Reports (Internet), 6, p.220 - 225, 2016/07
 C based on two binary non-ideal solid solutions
C based on two binary non-ideal solid solutionsWalker, C.; Suto, Shunkichi; Oda, Chie; Mihara, Morihiro; Honda, Akira
Cement and Concrete Research, 79, p.1 - 30, 2016/01
Times Cited Count:69 Percentile:90.65(Construction & Building Technology)Modeling the solubility behavior of calcium silicate hydrate (C-S-H) gel is important to make quantitative predictions of the degradation of hydrated ordinary Portland cement (OPC) based materials. Experimental C-S-H gel solubility data have been compiled from the literature, critically evaluated and supplemented with new data from the current study for molar Ca/Si ratios = 0.2-0.83. All these data have been used to derive a discrete solid phase (DSP) type C-S-H gel solubility model based on two binary non-ideal solid solutions in aqueous solution(SSAS). Features of the DSP type C-S-H gel solubility model include satisfactory predictions of pH values and Ca and Si concentrations for all molar Ca/Si ratios = 2.7  0 in the C-S-H system, portlandite (CH) for Ca/Si ratios
0 in the C-S-H system, portlandite (CH) for Ca/Si ratios 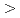 1.65, congruent dissolution at Ca/Si ratios = 0.85, and amorphous silica (SiO
1.65, congruent dissolution at Ca/Si ratios = 0.85, and amorphous silica (SiO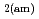 ) for Ca/Si ratios
) for Ca/Si ratios 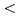 0.55 as identified in the current study by IR spectroscopy.
0.55 as identified in the current study by IR spectroscopy.
Kai, Takeshi; Yokoya, Akinari; Ukai, Masatoshi*; Fujii, Kentaro; Watanabe, Ritsuko
Radiation Physics and Chemistry, 115, p.1 - 5, 2015/10
Times Cited Count:34 Percentile:94.28(Chemistry, Physical)Role of secondary electrons on DNA damage have not been understood sufficiently because there still exists a lack of study for thermalization process of an electron in liquid phase. We calculated thermalization lengths and spatial distributions of an electron in liquid water using cross sections for rotation and phonon excitations in a liquid phase. Obtained thermalization lengths are in good agreement with experimental results reported by literatures. Thermalization time was also estimated from time evolution of spatial distributions of the incident electron to be hundreds femtoseconds. From these results, we predict that thermalization and pre-hydration of electron might progress simultaneously. These electrons possibly cause damage in biological molecules in a cell. Particularly severe types of DNA damage consisting of proximately located multiple lesions are potentially induced by reaction of DNA with the thermalized electrons by dissociative electron transfer.
Machida, Masahiko; Okumura, Masahiko; Nakamura, Hiroki; Sakuramoto, Kazuhiro*
Proceedings of Joint International Conference on Mathematics and Computation, Supercomputing in Nuclear Applications and the Monte Carlo Method (M&C + SNA + MC 2015) (CD-ROM), 12 Pages, 2015/04
In order to clarify physicochemical behaviors of radioactive Cs released into environment from the Fukushima Daiichi Nuclear Power Plants, we study on two issues, i.e., hydration structures of Cs and its adsorption on a specific edge in a clay particle (mica) by employing first principles calculations. In this paper, firstly, we report on hydration structures of Cs by using Born-Oppenheimer first-principles molecular dynamics. Our striking finding in the hydration structures is that Cs has no clear second hydration shell in contrast to any other alkali cations. Secondly, we construct a model of the Frayed Edge Site and confirm that the model actually becomes selective for Cs when expanding the interlayer distance from that of the original crystal structure through the calculation of the ion-exchange energy.
and its adsorption on a specific edge in a clay particle (mica) by employing first principles calculations. In this paper, firstly, we report on hydration structures of Cs by using Born-Oppenheimer first-principles molecular dynamics. Our striking finding in the hydration structures is that Cs has no clear second hydration shell in contrast to any other alkali cations. Secondly, we construct a model of the Frayed Edge Site and confirm that the model actually becomes selective for Cs when expanding the interlayer distance from that of the original crystal structure through the calculation of the ion-exchange energy.
Nagaishi, Ryuji; Inoue, Masao; Hino, Ryutaro; Ogawa, Toru
Proceedings of 2014 Nuclear Plant Chemistry Conference (NPC 2014) (USB Flash Drive), 9 Pages, 2014/10
Since seawater has been used as a coolant for reactors and spent fuel pools in broken reactor buildings at Fukushima Daiichi NPS accident, radioactive contaminated water emitted following the accident has contained salt content of seawater at high concentrations, different from that at TMI-2 accident. Radiolysis of seawater leading to hydrogen generation and corrosion has been simulated and reported by several groups. However, the proposed radiolysis models cannot be always applied to water radiolysis at the wide range of salt concentrations present in the NPS, mainly because primary yields of radiolysis products of water and radiation-induced reactions are dependent on the salt concentration. In this study, the radiolytic behavior in diluted and concentrated systems of seawater was considered on the basis of results in steady state and pulse radiolysis experiments, in which the above salt effects were demonstrated from the obtained results.
Maeda, Toshikatsu; Bamba, Tsunetaka*; Hotta, Katsutoshi*; Mizuno, Tsuyoshi*; Ozawa, Tatsuya
Nihon Genshiryoku Gakkai Wabun Rombunshi, 4(4), p.242 - 247, 2005/12
no abstracts in English
Kimura, Takaumi; Kirishima, Akira*; Arisaka, Makoto
Kidorui, (47), p.43 - 56, 2005/11
no abstracts in English
Jochi, Yasumasa*; Kitao, Akio*; Go, Nobuhiro
Journal of the American Chemical Society, 127(24), p.8705 - 8709, 2005/06
Times Cited Count:27 Percentile:61.19(Chemistry, Multidisciplinary)no abstracts in English
observed by neutron diffraction measurementsArai, Shigeki; Chatake, Toshiyuki*; Ohara, Takashi; Kurihara, Kazuo; Tanaka, Ichiro*; Suzuki, Nobuhiro*; Fujimoto, Zui*; Mizuno, Hiroshi*; Niimura, Nobuo
Nucleic Acids Research, 33(9), p.3017 - 3024, 2005/05
Times Cited Count:93 Percentile:82.96(Biochemistry & Molecular Biology)It has long been suspected that the structure and function of a DNA duplex can be strongly dependent on its degree of hydration. By neutron diffraction experiments, we have succeeded in determining most of the hydrogen (H) and deuterium (D) atomic positions in the d(CCATTAATGG) duplex. Moreover, the D positions in 27 DO molecules have been determined. In particular, the complex water network in the minor groove has been observed in detail. By a combined structural analysis using 2.0 Å resolution X-ray and 3.0 Å resolution neutron data, it is clear that the spine of hydration is built up, not only by a simple hexagonal hydration pattern (as reported in prior X-ray studies), but also by many other water bridges hydrogen-bonded to the DNA strands. The complexity of the hydration pattern in the minor groove is derived from an extraordinary variety of orientations displayed by the water molecules.
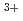 ion; A Car-Parrinello molecular dynamics study
ion; A Car-Parrinello molecular dynamics studyIkeda, Takashi; Hirata, Masaru; Kimura, Takaumi
Journal of Chemical Physics, 122(2), p.024510_1 - 024510_5, 2005/01
Times Cited Count:31 Percentile:68.38(Chemistry, Physical)The solvation shell structure of Y and the dynamics of the hydrated ion in an aqueous solution of 0.8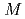 YCl are studied in two conditions with and without an excess proton by using first principles molecular dynamics method. We find that the first solvation shell around Y contains eight water molecules forming a square antiprism as expected from X-ray absorption near edge structure in both the conditions we examined. A detailed analysis relying upon localized orbitals reveals that the complexation of water molecules with yttrium cation leads to a substantial amount of charge redistribution particularly on the oxygen atoms, giving rise to the chemical shifts of
YCl are studied in two conditions with and without an excess proton by using first principles molecular dynamics method. We find that the first solvation shell around Y contains eight water molecules forming a square antiprism as expected from X-ray absorption near edge structure in both the conditions we examined. A detailed analysis relying upon localized orbitals reveals that the complexation of water molecules with yttrium cation leads to a substantial amount of charge redistribution particularly on the oxygen atoms, giving rise to the chemical shifts of  -20 ppm in
-20 ppm in 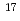 O NMR relative to the computed nuclear shieldings of the bulk water.
O NMR relative to the computed nuclear shieldings of the bulk water.
Yoshida, Yoichi*; Yang, J.*; Saeki, Akinori*; Tagawa, Seiichi*; Shibata, Hiromi*; Namba, Hideki; Kojima, Takuji; Taguchi, Mitsumasa
JAERI-Review 2004-025, TIARA Annual Report 2003, p.143 - 144, 2004/11
no abstracts in English
Zhang, P.*; Kimura, Takaumi; Yoshida, Zenko
Solvent Extraction and Ion Exchange, 22(6), p.933 - 945, 2004/00
Times Cited Count:13 Percentile:45.29(Chemistry, Multidisciplinary)no abstracts in English
